
Relevant publications
Bioinformatics & Computational Chemistry
A theoretical framework for an efficient normalizing flow-based solution to the Schrödinger equation

A central problem in quantum mechanics involves solving the Electronic Schrödinger Equation for a molecule or material. The Variational Monte Carlo approach to this problem approximates a particular variational objective via sampling, and then optimizes this approximated objective over a chosen parameterized family of wavefunctions, known as the ansatz. Recently neural networks have been used as the ansatz, with accompanying success. However, sampling from such wavefunctions has required the use of a Markov Chain Monte Carlo approach, which is inherently inefficient. In this work, we propose a solution to this problem via an ansatz which is cheap to sample from, yet satisfies the requisite quantum mechanical properties. We prove that a normalizing flow using the following two essential ingredients satisfies our requirements: (a) a base distribution which is constructed from Determinantal Point Processes; (b) flow layers which are equivariant to a particular subgroup of the permutation group. We then show how to construct both continuous and discrete normalizing flows which satisfy the requisite equivariance. We further demonstrate the manner in which the non-smooth nature (“cusps”) of the wavefunction may be captured, and how the framework may be generalized to provide induction across multiple molecules. The resulting theoretical framework entails an efficient approach to solving the Electronic Schrödinger Equation.
Inverse problems with experiment-guided AlphaFold
Proteins exist as a dynamic ensemble of multiple conformations, and these motions are often crucial for their functions. However, current structure prediction methods predominantly yield a single conformation, overlooking the conformational heterogeneity revealed by diverse experimental modalities. Here, we present a framework for building experiment-grounded protein structure generative models that infer conformational ensembles consistent with measured experimental data. The key idea is to treat state-of-the-art protein structure predictors (e.g., AlphaFold3) as sequence-conditioned structural priors, and cast ensemble modeling as posterior inference of protein structures given experimental measurements. Through extensive real-data experiments, we demonstrate the generality of our method to incorporate a variety of experimental measurements. In particular, our framework uncovers previously unmodeled conformational heterogeneity from crystallographic densities, and generates high-accuracy NMR ensembles orders of magnitude faster than the status quo. Notably, we demonstrate that our ensembles outperform AlphaFold3 and sometimes better fit experimental data than publicly deposited structures to the Protein Data Bank (PDB). We believe that this approach will unlock building predictive models that fully embrace experimentally observed conformational diversity.
How local is "local"? Deep learning reveals locality of the induced magnetic field of polycyclic aromatic hydrocarbons
We investigate the locality of magnetic response in polycyclic aromatic molecules using a novel deep-learning approach. Our method employs graph neural networks (GNNs) with a graph-of-rings representation to predict Nucleus-Independent Chemical Shifts in the space around the molecule. We train a series of models, each time reducing the size of the largest molecules used in training. The accuracy of prediction remains high (MAE < 0.5 ppm), even when training the model only on molecules with up to 4 rings, thus providing strong evidence for the locality of magnetic response. To overcome the known problem of generalization of GNNs, we implement a k-hop expansion strategy and succeed in achieving accurate predictions for molecules with up to 15 rings (almost 4 times the size of the largest training example). Our findings have implications for understanding the magnetic response in complex molecules and demonstrate a promising approach to overcoming GNN scalability limitations. Furthermore, the trained models enable rapid characterization, without the need for more expensive DFT calculations.
Generative modeling of protein ensembles guided by crystallographic electron densities
Proteins are dynamic, adopting ensembles of conformations. The nature of this conformational heterogenity is imprinted in the raw electron density measurements obtained from X-ray crystallography experiments. Fitting an ensemble of protein structures to these measurements is a challenging, ill-posed inverse problem. We propose a non-i.i.d. ensemble guidance approach to solve this problem using existing protein structure generative models and demonstrate that it accurately recovers complicated multi-modal alternate protein backbone conformations observed in certain single crystal measurements.
Beyond the alphabet: deep signal embedding for enhanced DNA clustering
The emerging field of DNA storage employs strands of DNA bases (A/T/C/G) as a storage medium for digital information to enable massive density and durability. The DNA storage pipeline includes: (1) encoding the raw data into sequences of DNA bases; (2) synthesizing the sequences as DNA strands that are stored over time as an unordered set; (3) sequencing the DNA strands to generate DNA reads; and (4) deducing the original data. The DNA synthesis and sequencing stages each generate several independent error-prone duplicates of each strand which are then utilized in the final stage to reconstruct the best estimate for the original strand. Specifically, the reads are first clustered into groups likely originating from the same strand (based on their similarity to each other), and then each group approximates the strand that led to the reads of that group. This work improves the DNA clustering stage by embedding it as part of the DNA sequencing. Traditional DNA storage solutions begin after the DNA sequencing process generates discrete DNA reads (A/T/C/G), yet we identify that there is untapped potential in using the raw signals generated by the Nanopore DNA sequencing machine before they are discretized into bases, a process known as basecalling, which is done using a deep neural network. We propose a deep neural network that clusters these signals directly, demonstrating superior accuracy, and reduced computation times compared to current approaches that cluster after basecalling.
Seeing Double: Molecular dynamics simulations reveal the stability of certain alternate protein conformations in crystal structures
Proteins jiggle around, adopting ensembles of interchanging conformations. Here we show through a large-scale analysis of the Protein Data Bank and using molecular dynamics simulations, that segments of protein chains can also commonly adopt dual, transiently stable conformations which is not explained by direct interactions. Our analysis highlights how alternate conformations can be maintained as non-interchanging, separated states intrinsic to the protein chain, namely through steric barriers or the adoption of transient secondary structure elements. We further demonstrate that despite the commonality of the phenomenon, current structural ensemble prediction methods fail to capture these bimodal distributions of conformations.
A dataset of alternately located segments in protein crystal structures
Protein Data Bank (PDB) files list the relative spatial location of atoms in a protein structure as the final output of the process of fitting and refining to experimentally determined electron density measurements. Where experimental evidence exists for multiple conformations, atoms are modelled in alternate locations. Programs reading PDB files commonly ignore these alternate conformations by default leaving users oblivious to the presence of alternate conformations in the structures they analyze. This has led to underappreciation of their prevalence, under characterisation of their features and limited the accessibility to this high-resolution data representing structural ensembles. We have trawled PDB files to extract structural features of residues with alternately located atoms. The output includes the distance between alternate conformations and identifies the location of these segments within the protein chain and in proximity of all other atoms within a defined radius. This dataset should be of use in efforts to predict multiple structures from a single sequence and support studies investigating protein flexibility and the association with protein function.
An amino-domino model described by a cross-peptide-bond Ramachandran plot defines amino acid pairs as local structural units
Protein structure, both at the global and local level, dictates function. Proteins fold from chains of amino acids, forming secondary structures, α-helices and β-strands, that, at least for globular proteins, subsequently fold into a three-dimensional structure. Here, we show that a Ramachandran-type plot focusing on the two dihedral angles separated by the peptide bond, and entirely contained within an amino acid pair, defines a local structural unit. We further demonstrate the usefulness of this cross-peptide-bond Ramachandran plot by showing that it captures β-turn conformations in coil regions, that traditional Ramachandran plot outliers fall into occupied regions of our plot, and that thermophilic proteins prefer specific amino acid pair conformations. Further, we demonstrate experimentally that the effect of a point mutation on backbone conformation and protein stability depends on the amino acid pair context, i.e., the identity of the adjacent amino acid, in a manner predictable by our method.
Guided diffusion for inverse molecular design
The holy grail of materials science is de novo molecular design — i.e., the ability to engineer molecules with desired characteristics. Recently, this goal has become increasingly achievable thanks to developments such as equivariant graph neural networks that can better predict molecular properties, and to the improved performance of generation tasks, in particular of conditional generation, in text-to-image generators and large language models. Herein, we introduce GaUDI, a guided diffusion model for inverse molecular design, which combines these advances and can generate novel molecules with desired properties. GaUDI decouples the generator and the property-predicting models and can be guided using both point-wise targets and open-ended targets (e.g., minimum/maximum). We demonstrate GaUDI’s effectiveness using single- and multiple-objective tasks applied to newly-generated data sets of polycyclic aromatic systems, achieving nearly 100% validity of generated molecules. Further, for some tasks, GaUDI discovers better molecules than those present in our data set of 475k molecules.
Interpretable deep learning unveils structure-property relationships in polybenzenoid hydrocarbons
In this work, interpretable deep learning was used to identify structure-property relationships governing the HOMO-LUMO gap and relative stability of polybenzenoid hydrocarbons (PBHs). To this end, a ring-based graph representation was used. In addition to affording reduced training times and excellent predictive ability, this representation could be combined with a subunit-based perception of PBHs, allowing chemical insights to be presented in terms of intuitive and simple structural motifs. The resulting insights agree with conventional organic chemistry knowledge and electronic structure-based analyses, and also reveal new behaviors and identify influential structural motifs. In particular, we evaluated and compared the effects of linear, angular, and branching motifs on these two molecular properties, as well as explored the role of dispersion in mitigating torsional strain inherent in non-planar PBHs. Hence, the observed regularities and the proposed analysis contribute to a deeper understanding of the behavior of PBHs and form the foundation for design strategies for new functional PBHs.
Water stabilizes an alternate turn conformation in horse heart myoglobin
Comparison of myoglobin structures reveals that protein isolated from horse heart consistently adopts an alternate turn conformation in comparison to its homologues. Analysis of hundreds of high-resolution structures discounts crystallization conditions or the surrounding amino acid protein environment as explaining this difference, that is also not captured by the AlphaFold prediction. Rather, a water molecule is identified as stabilizing the conformation in the horse heart structure, which immediately reverts to the whale conformation in molecular dynamics simulations excluding that structural water.
Machine learning approaches demonstrate that protein structures carry information about their genetic coding
Synonymous codons translate into the same amino acid. Although the identity of synonymous codons is often considered
inconsequential to the final protein structure there is mounting evidence for an association between the two. Our study
examined this association using regression and classification models, finding that codon sequences predict protein backbone dihedral angles with a lower error than amino acid sequences, and that models trained with true dihedral angles have better classification of synonymous codons given structural information than models trained with random dihedral angles. Using this classification approach, we investigated local codon-codon dependencies and tested whether synonymous codon identity can be predicted more accurately from codon context than amino acid context alone, and most specifically which codon context position carries the most predictive power.
Defining amino acid pairs as structural units suggests mutation sensitivity to adjacent residues
Proteins fold from chains of amino acids, forming secondary structures, α-helices and β-strands, that, at least for globular proteins, subsequently fold into a three-dimensional structure. A large-scale analysis of high-resolution protein structures suggests that amino acid pairs constitute another layer of ordered structure, more local than these conventionally defined secondary structures. We develop a cross-peptide-bond Ramachandran plot that captures the 15 conformational preferences of the amino acid pairs and show that the effect of a particular mutation on the stability of a protein depends in a predictable manner on the adjacent amino acid context.
Codon-specific Ramachandran plots show amino acid backbone conformation depends on identity of the translated codon
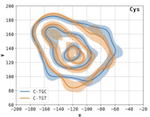
Synonymous codons translate into chemically identical amino acids. Once considered inconsequential to the formation of the protein product, there is now significant evidence to suggest that codon usage affects co-translational protein folding and the final structure of the expressed protein. Here we develop a method for computing and comparing codon-specific Ramachandran plots and demonstrate that the backbone dihedral angle distributions of some synonymous codons are distinguishable with statistical significance for some secondary structures. This shows that there exists a dependence between codon identity and backbone torsion of the translated amino acid. Although these findings cannot pinpoint the causal direction of this dependence, we discuss the vast biological implications should coding be shown to directly shape protein conformation and demonstrate the usefulness of this method as a tool for probing associations between codon usage and protein structure. Finally, we urge for the inclusion of exact genetic information into structural databases.