
Relevant publications
Dr. Aviv Rosenberg
Seeing Double: Molecular dynamics simulations reveal the stability of certain alternate protein conformations in crystal structures

Proteins jiggle around, adopting ensembles of interchanging conformations. Here we show through a large-scale analysis of the Protein Data Bank and using molecular dynamics simulations, that segments of protein chains can also commonly adopt dual, transiently stable conformations which is not explained by direct interactions. Our analysis highlights how alternate conformations can be maintained as non-interchanging, separated states intrinsic to the protein chain, namely through steric barriers or the adoption of transient secondary structure elements. We further demonstrate that despite the commonality of the phenomenon, current structural ensemble prediction methods fail to capture these bimodal distributions of conformations.
A dataset of alternately located segments in protein crystal structures
Protein Data Bank (PDB) files list the relative spatial location of atoms in a protein structure as the final output of the process of fitting and refining to experimentally determined electron density measurements. Where experimental evidence exists for multiple conformations, atoms are modelled in alternate locations. Programs reading PDB files commonly ignore these alternate conformations by default leaving users oblivious to the presence of alternate conformations in the structures they analyze. This has led to underappreciation of their prevalence, under characterisation of their features and limited the accessibility to this high-resolution data representing structural ensembles. We have trawled PDB files to extract structural features of residues with alternately located atoms. The output includes the distance between alternate conformations and identifies the location of these segments within the protein chain and in proximity of all other atoms within a defined radius. This dataset should be of use in efforts to predict multiple structures from a single sequence and support studies investigating protein flexibility and the association with protein function.
Vector quantile regression on manifolds
Quantile regression (QR) is a statistical tool for distribution-free estimation of conditional quantiles of a target variable given explanatory features. QR is limited by the assumption that the target distribution is univariate and defined on an Euclidean domain. Although the notion of quantiles was recently extended to multi-variate distributions, QR for multi-variate distributions on manifolds remains underexplored, even though many important applications inherently involve data distributed on, e.g., spheres (climate and geological phenomena), and tori (dihedral angles in proteins). By leveraging optimal transport theory and c-concave functions, we meaningfully define conditional vector quantile functions of high-dimensional variables on manifolds (M-CVQFs). Our approach allows for quantile estimation, regression, and computation of conditional confidence sets and likelihoods. We demonstrate the approach’s efficacy and provide insights regarding the meaning of non-Euclidean quantiles through synthetic and real data experiments.
An amino-domino model described by a cross-peptide-bond Ramachandran plot defines amino acid pairs as local structural units
Protein structure, both at the global and local level, dictates function. Proteins fold from chains of amino acids, forming secondary structures, α-helices and β-strands, that, at least for globular proteins, subsequently fold into a three-dimensional structure. Here, we show that a Ramachandran-type plot focusing on the two dihedral angles separated by the peptide bond, and entirely contained within an amino acid pair, defines a local structural unit. We further demonstrate the usefulness of this cross-peptide-bond Ramachandran plot by showing that it captures β-turn conformations in coil regions, that traditional Ramachandran plot outliers fall into occupied regions of our plot, and that thermophilic proteins prefer specific amino acid pair conformations. Further, we demonstrate experimentally that the effect of a point mutation on backbone conformation and protein stability depends on the amino acid pair context, i.e., the identity of the adjacent amino acid, in a manner predictable by our method.
Continuous vector quantile regression
Vector quantile regression (VQR) estimates the conditional vector quantile function (CVQF), a fundamental quantity which fully represents the conditional distribution of Y|X. VQR is formulated as an optimal transport (OT) problem between a uniform U~μ and the target (X,Y)~ν, the solution of which is a unique transport map, co-monotonic with U. Recently NL-VQR has been proposed to estimate support non-linear CVQFs, together with fast solvers which enabled the use of this tool in practical applications. Despite its utility, the scalability and estimation quality of NL-VQR is limited due to a discretization of the OT problem onto a grid of quantile levels. We propose a novel continuous formulation and parametrization of VQR using partial input-convex neural networks (PICNNs). Our approach allows for accurate, scalable, differentiable and invertible estimation of non-linear CVQFs. We further demonstrate, theoretically and experimentally, how continuous CVQFs can be used for general statistical inference tasks: estimation of likelihoods, CDFs, confidence sets, coverage, sampling, and more. This work is an important step towards unlocking the full potential of VQR.
Vector quantile regression on manifolds
Quantile regression (QR) is a statistical tool for distribution-free
estimation of conditional quantiles of a target variable given explanatory
features. QR is limited by the assumption that the target distribution is
univariate and defined on an Euclidean domain. Although the notion of quantiles
was recently extended to multi-variate distributions, QR for multi-variate
distributions on manifolds remains underexplored, even though many important
applications inherently involve data distributed on, e.g., spheres (climate
measurements), tori (dihedral angles in proteins), or Lie groups (attitude in
navigation). By leveraging optimal transport theory and the notion of
c-concave functions, we meaningfully define conditional vector quantile
functions of high-dimensional variables on manifolds (M-CVQFs). Our approach
allows for quantile estimation, regression, and computation of conditional
confidence sets. We demonstrate the approach’s efficacy and provide insights
regarding the meaning of non-Euclidean quantiles through preliminary synthetic
data experiments.
Fast nonlinear vector quantile regression
Quantile regression (QR) is a powerful tool for estimating one or more conditional quantiles of a target variable Y given explanatory features X. A limitation of QR is that it is only defined for scalar target variables, due to the formulation of its objective function, and since the notion of quantiles has no standard definition for multivariate distributions. Recently, vector quantile regression (VQR) was proposed as an extension of QR for high-dimensional target variables, thanks to a meaningful generalization of the notion of quantiles to multivariate distributions. Despite its elegance, VQR is arguably not applicable in practice due to several limitations: (i) it assumes a linear model for the quantiles of the target Y given the features X; (ii) its exact formulation is intractable even for modestly-sized problems in terms of target dimensions, the number of regressed quantile levels, or the number of features, and its relaxed dual formulation may violate the monotonicity of the estimated quantiles; (iii) no fast or scalable solvers for VQR currently exist. In this work we fully address these limitations, namely: (i) We extend VQR to the non-linear case, showing substantial improvement over linear VQR; (ii) We propose vector monotone rearrangement, a method which ensures the estimates obtained by VQR relaxations are monotone functions; (iii) We provide fast, GPU-accelerated solvers for linear and nonlinear VQR which maintain a fixed memory footprint with the number of samples and quantile levels, and demonstrate that they scale to millions of samples and thousands of quantile levels; (iv) We release an optimized python package of our solvers as to widespread the use of VQR in real-world applications.
Machine learning approaches demonstrate that protein structures carry information about their genetic coding
Synonymous codons translate into the same amino acid. Although the identity of synonymous codons is often considered
inconsequential to the final protein structure there is mounting evidence for an association between the two. Our study
examined this association using regression and classification models, finding that codon sequences predict protein backbone dihedral angles with a lower error than amino acid sequences, and that models trained with true dihedral angles have better classification of synonymous codons given structural information than models trained with random dihedral angles. Using this classification approach, we investigated local codon-codon dependencies and tested whether synonymous codon identity can be predicted more accurately from codon context than amino acid context alone, and most specifically which codon context position carries the most predictive power.
Defining amino acid pairs as structural units suggests mutation sensitivity to adjacent residues
Proteins fold from chains of amino acids, forming secondary structures, α-helices and β-strands, that, at least for globular proteins, subsequently fold into a three-dimensional structure. A large-scale analysis of high-resolution protein structures suggests that amino acid pairs constitute another layer of ordered structure, more local than these conventionally defined secondary structures. We develop a cross-peptide-bond Ramachandran plot that captures the 15 conformational preferences of the amino acid pairs and show that the effect of a particular mutation on the stability of a protein depends in a predictable manner on the adjacent amino acid context.
Codon-specific Ramachandran plots show amino acid backbone conformation depends on identity of the translated codon
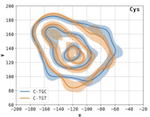
Synonymous codons translate into chemically identical amino acids. Once considered inconsequential to the formation of the protein product, there is now significant evidence to suggest that codon usage affects co-translational protein folding and the final structure of the expressed protein. Here we develop a method for computing and comparing codon-specific Ramachandran plots and demonstrate that the backbone dihedral angle distributions of some synonymous codons are distinguishable with statistical significance for some secondary structures. This shows that there exists a dependence between codon identity and backbone torsion of the translated amino acid. Although these findings cannot pinpoint the causal direction of this dependence, we discuss the vast biological implications should coding be shown to directly shape protein conformation and demonstrate the usefulness of this method as a tool for probing associations between codon usage and protein structure. Finally, we urge for the inclusion of exact genetic information into structural databases.
Meeting the unmet needs of clinicians from AI systems showcased for cardiology with deep-learning-based ECG analysis
Despite their great promise, artificial intelligence (AI) systems have yet to become ubiquitous in the daily practice of medicine largely due to several crucial unmet needs of healthcare practitioners. These include lack of explanations in clinically meaningful terms, handling the presence of unknown medical conditions, and transparency regarding the system’s limitations, both in terms of statistical performance as well as recognizing situations for which the system’s predictions are irrelevant. We articulate these unmet clinical needs as machine-learning (ML) problems and systematically address them with cutting-edge ML techniques. We focus on electrocardiogram (ECG) analysis as an example domain in which AI has great potential and tackle two challenging tasks: the detection of a heterogeneous mix of known and unknown arrhythmias from ECG and the identification of underlying cardio-pathology from segments annotated as normal sinus rhythm recorded in patients with an intermittent arrhythmia. We validate our methods by simulating a screening for arrhythmias in a large-scale population while adhering to statistical significance requirements. Specifically, our system 1) visualizes the relative importance of each part of an ECG segment for the final model decision; 2) upholds specified statistical constraints on its out-of-sample performance and provides uncertainty estimation for its predictions; 3) handles inputs containing unknown rhythm types; and 4) handles data from unseen patients while also flagging cases in which the model’s outputs are not usable for a specific patient. This work represents a significant step toward overcoming the limitations currently impeding the integration of AI into clinical practice in cardiology and medicine in general.